
What is GSPR (General Safety And Performance Requirements) for EU MDR and EU IVDR?
Establish conformity with these requirements and should provide sufficient evidence to demonstrate compliance with GSPR. GSPR is a core element to navigate CE marking for a device.
By Soumya Mahapatra
December 16, 2021

GSPR Chapters and Requirements Guide:
EU MDR 2017/745EU IVDR 2017/746Chapter 1 – General requirements (1 to 9)Chapter 1 – General requirements (from 1 to 8)Chapter 2 -Design and Manuf. (from 10 to 22)Chapter 2 – Performance, Design and Manuf. (from 9 to 19)Chapter 3 –Labels and IFU (23)Chapter 3 – Labels and IFU (20)
Essential Requirements for EU MDD and EU IVDD
The Medical Device Directive (MDD) defines the “Essential Requirements”, as the requirements that every medical product has to fulfill, according to the scope they belong to. These essential requirements are described by Directive in Annex I, 93/42/EEC for EU MDD and 98/79/EEC for EU IVDD, and there are in total 13 requirements.
Essential Requirements Chapters and Requirements Guide:
EU MDD 93/42/EECEU IVDD 98/79/EECChapter 1 – General requirements (1 to 6)Chapter 1 –General requirements (from 1 to 5)Chapter 2 -Design and Manuf. (from 7 to 13)Chapter 2 -Performance, Design and Manuf. (from 1 to 8)
Layout and Structure of the GSPR
The General Safety and Performance Requirements (GSPR) are divided into the following 3 chapters:
1. General requirements
Intended purpose, safety of patients, users and other persons
Reduction of risks
Risk management system
Risk control measures
Risks related to use error
Device performance shall not be adversely affected
Device design, manufacture, packaging
Risk reduction, acceptable risk-benefit ratio
General safety requirements for devices without an intended medical purpose as described in in annex XVI
2. Requirements regarding design and manufacture
Chemical, physical and biological properties
Infection and microbial contamination
Devices incorporating a substance considered to be a medicinal product and devices that are composed of substances/substance combinations that are absorbed by or locally dispersed in the human body
Devices incorporating materials of biological origin
Construction of devices and interaction with their environment
Devices with a diagnostic and measuring function
Protection against radiation
Electronic programmable systems – devices that incorporate electronic programmable systems and software that are devices in themselves
Active devices and devices connected to them
Particular requirements for active implantable devices
Protection against mechanical and thermal risks
Protection against the risks posed to the patient or user by devices supplying energy or substances
Protection against the risks posed by medical devices intended by the manufacturer for use by lay persons
3. Requirements regarding the information supplied with the device
Label and instructions for use
Essential Requirements vs GSPR
EU MDR and EU IVDR replace the EU MDD and EU IVDD for CE Mark in Europe. As shown in the table above, the Essential Requirements of the MDD are divided into 2 chapters with 13 items while MDR has 3 chapters with 23 items. This means that the changes from MDD have not only shifted quite a bit, but the scope of the details in requirements has also increased considerably with MDR.
To understand the extent of changes, items covering “Information provided by the manufacturer” from Chapter 2 in Essential Requirements have now been reorganized and incorporated into a new chapter 3 in GSPR.
Additional requirements and some administrative simplification for MDR come from the fact that the MDD has been combined with AIMDD (Active Implantable Medical Device Directive ER) and is now covered within GSPR Item 19. Further, in MDR’s GSPR a number of topics have been given greater emphasis or have been dealt with in more detail.
Besides expanding on some key aspects, several topics are newly incorporated in the requirements list of the GSPR. These include but are not limited to:
Requirements for devices that administer (GSPR 10.3/4) or contain drugs (GSPR Item 12)
Specific requirements for devices that contain tissues of human or animal origin (GSPR 13)
Requirements for disposal (GSPR 14.7/23.4)
Requirements for IT safety (GSPR 17.4)
Requirements for devices for use by lay persons (GSPR 22)
General requirements for labelling (GSPR 23)
Chapter 1 of GSPR
Chapter 1 in GSPR has many similarities with the MDD. However, there is a higher emphasis on usability and dependability along the product life cycle and the state of the art. Paragraphs 2 – 5 of Chapter 1 stress on risk management and the importance of medical devices. While paragraph 9 of Chapter 1 addresses devices without an intended medical purpose, the other paragraphs are very similar to the MDD and depict the “standard” requirements for every medical device.
Chapter 2 of GSPR
Key highlights as it pertains to changes from chapter 2 of GSPR are outlined below:
Paragraph 10 provides more details for the chemical, physical and biological properties, especially as it relates to handling of toxicity and specific substances
Paragraph 11 addresses requirements on infection and microbial contamination
The scope in paragraph 12 is extended to include substances absorbed or locally dispersed by the human body
Paragraph 13 now includes non-viable human tissue in the biological tissue category
New regulations have also been added for the interaction of medical devices with the environment (paragraph 14) and the compatibility with other devices
Network and cybersecurity has grown in importance within the global medical device diaspora and as such they are given high importance in the GSPR
Regulations for mechanical and thermal risks as well as risk reduction have also become more detailed
Chapter 3 of GSPR
In chapter 3 of GSPR, paragraph 23 “label and instructions for use” is covered in detail. In general, this chapter handles significantly more requirements, such as the format of the instructions for use, readability, comprehensibility, availability, and how this relates with laypersons in addition to professionals. This chapter also covers additional requirements that must be fulfilled for UDI labeling as well as devices containing human or animal tissue. The same applies to the labeling of sterile packaging or the indication of Carcinogenic Mutagenic and toxic to Reproduction (CMR) substances.
Transitioning from MDD to MDR and from IVDD to IVDR
From 26th May 2020 to 25th May 2024, CE mark certificates issued under the MDD, before the MDR fully applies, will be valid for up to 4 years. From 25th May 2022 to 25th May 2024, CE mark certificates issued under the IVDD before the IVDR fully applies may remain valid for up to 2 additional years. From 26th May 2024, all devices placed on the market must be in conformity with the MDR or IVDR.
Below is an infographic showing the transition timeline for medical device manufacturers in Europe:
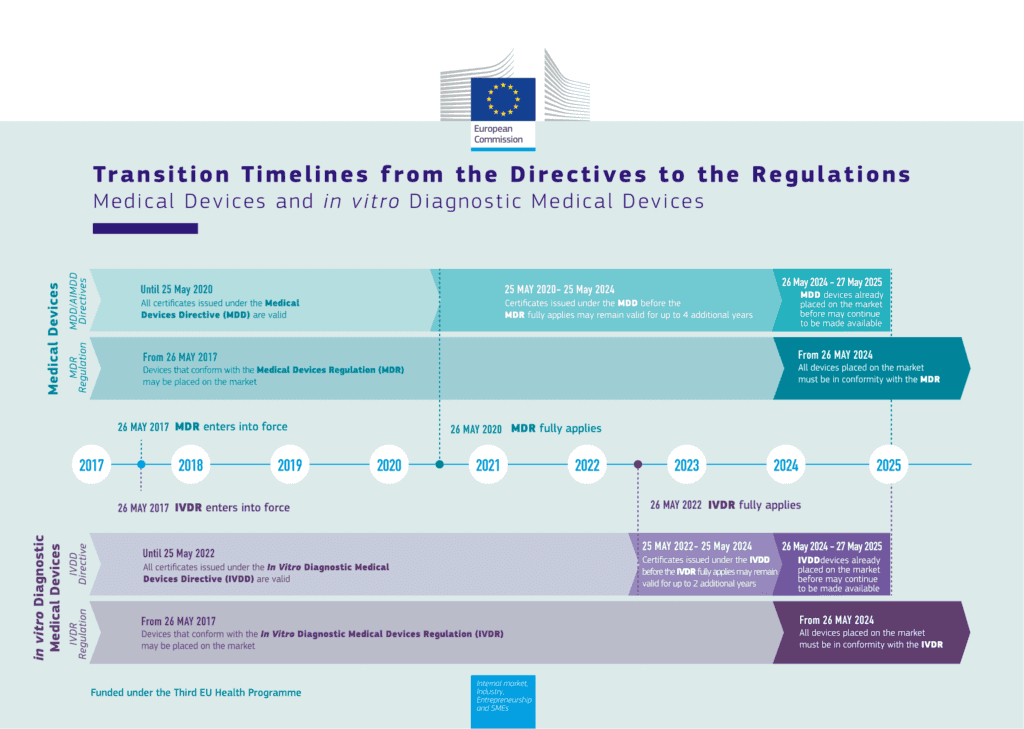
MDR and IVDR Transition Timeline
Challenges of Maintaining GSPR
From a regulatory point of view, there has been no fundamental shift with the introduction of GSPR. It is still crucial that the safety and performance of medical devices are proven. This must be acceptable within the given clinical context and the manufacturer needs to ensure that the two components do not change significantly during the entire life cycle of the medical device. That is why a systematic risk management process is required, which must be updated constantly, even after the device is in the market.
Yet another challenge with the GSPR lies with the fact that many state-of-the-art requirements from harmonized standards have been incorporated directly into the MDR. However, no annex from the harmonized standards refers to the GSPR in the MDR currently. Meaning, manufacturers will still need to perform a careful gap analysis in order to establish the relation until no uniformity exists between the two.
The objective evidence that the GSPR is fulfilled is part of the Technical File. Also, during the selected conformity assessment procedure, it must be proven that the requirements in GSPR are fulfilled.
Best Practices to Streamline GSPR For your Medical Devices
GSPR as such is not just another document to be updated, but rather a critical artifact that guides the development process and decisions from the outset for a manufacturer. Therefore, medical device manufacturers should promptly prepare a new checklist for the General Safety and Performance Requirements (GSPR) according to Annex I of MDR and IVDR to ensure the new GSPR is implemented and complied timely.
Using the GSPR checklist is practical and ensures traceability and completeness, but the number of items and the ongoing process of adding references to evidence and methods of conformity in the form of searching and linking 100s of standards, evidence documents, or procedures especially as they change and evolve requires the constant upkeep and maintenance of GSPR for products.
Essenvia’s Regulatory Management tool allows you to easily maintain GSPR for your products and variants. It serves as a single source of truth that links to standards databases and evidence documents and reports within the platform, helping you easily cross-reference methods of conformity whether these are harmonized standards, procedures from your quality system, or testing reports. Easily create, publish and link new versions of the GSPR with the Technical File to maintain compliance with Annex I requirements for the MDR. Schedule a demo now to find out how Essenvia can help you with your regulatory submissions.