
What do Regulatory Specialists need to know about searching the FDA 510(k) Database for Regulatory Strategy
Medical device manufacturers who intend to introduce a device into commercial distribution in the United States must submit a premarket notification – 510(k) or PMN to the FDA.This rule is applicable whether you are introducing a device for the first time, or planning a reintroduction with significant modifications.
By Soumya Mahapatra
September 20, 2021

Overview
Medical device manufacturers who intend to introduce a device into commercial distribution in the United States must submit a premarket notification – 510(k) or PMN to the FDA.This rule is applicable whether you are introducing a device for the first time, or planning a reintroduction with significant modifications. To ensure this rule is followed, you must leverage the information found in the FDA 510 (k) database.
This regulatory submission allows the FDA to determine whether the device is equivalent to a device already placed into one of the three classification categories. These categories include: Class I or Class II- with or without exemptions or Class III Premarket Approval (PMA).
If your device is classified as Class I or II, and if it is not exempt, a 510(k) will be required for marketing.
510(k) Submission Pathway:
A 510(k) is a premarket submission that needs to be sent to the FDA to demonstrate that the device to be marketed is safe and effective. It will also prove the device is substantially equivalent, to a legally marketed device (section 513(i)(1)(A) FD&C Act).
Once the device is determined to be substantially equivalent and cleared by FDA it can then be marketed in the U.S. The substantially equivalent determination is usually made within 90 days. This determination is based on the information submitted by medical device company seeking the clearance.
However, before you decide whether your device is eligible for the 510(k) pathway it is critical that you understand the regulatory strategy for your device.
The important elements of a regulatory strategy include:
FDA Product Code
Regulation Number
Proposed Indications For Use
Testing requirements
Product and device-specific guidance documents
In the paragraphs to follow we explain how the FDA 510(k) database is the ideal resource for medical device manufacturers to assess and determine their regulatory strategy to market.
How to use the FDA 510(k) Database to assess your regulatory strategy:
FDA maintains a database of previously cleared 510(k)s on its website. Typically, they will add new devices around the 5th of each month for devices cleared in the prior month. Users can search for previously cleared 510(k) submissions from this database using search criteria such as but not limited to:
510(k) Number
Applicant Name
Product Code
Device name
Steps to performing an efficient 510(k) database search:
The following steps outline how to use the FDA 510(k) database to create your regulatory strategy and manage ongoing updates:
Prepare your 510(k) search criteria:
For creating an effective regulatory strategy it is extremely important to take a methodical approach. The first step, therefore, is to document the technological characteristics, the intended use, and the desired indications of use for your device. Also, identify intended users of your product. This may include patients, caregivers, or healthcare professionals.
Build your initial keywords list based on the important attributes describe the technology (for example a needle, a catheter, etc.). This key word list also includes the indications and mode of action for the device. These keywords are attributes that when combined represent your device.
Identify and list your competitors and the devices they have marketed or received approval for. We will talk about how to use this information later in this article.
Perform a search of the 510(k) database
There are two ways to run an efficient search on the FDA 510(k) database: (1) Quick search and (2) Advanced search. We will discuss how to leverage each of these search methods to optimize this FDA resource.A quick search on the 510(k) database with your keywords is helpful to identify a list of devices that match your device category.Below is an example of how this search works using an apnea monitor as the device
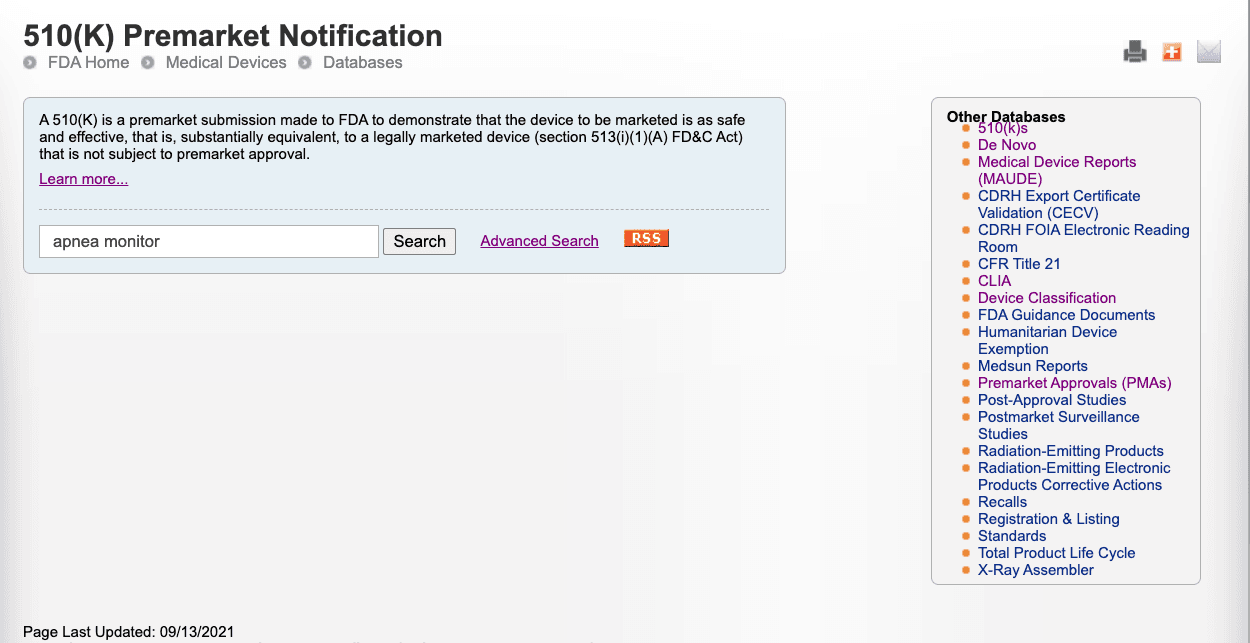
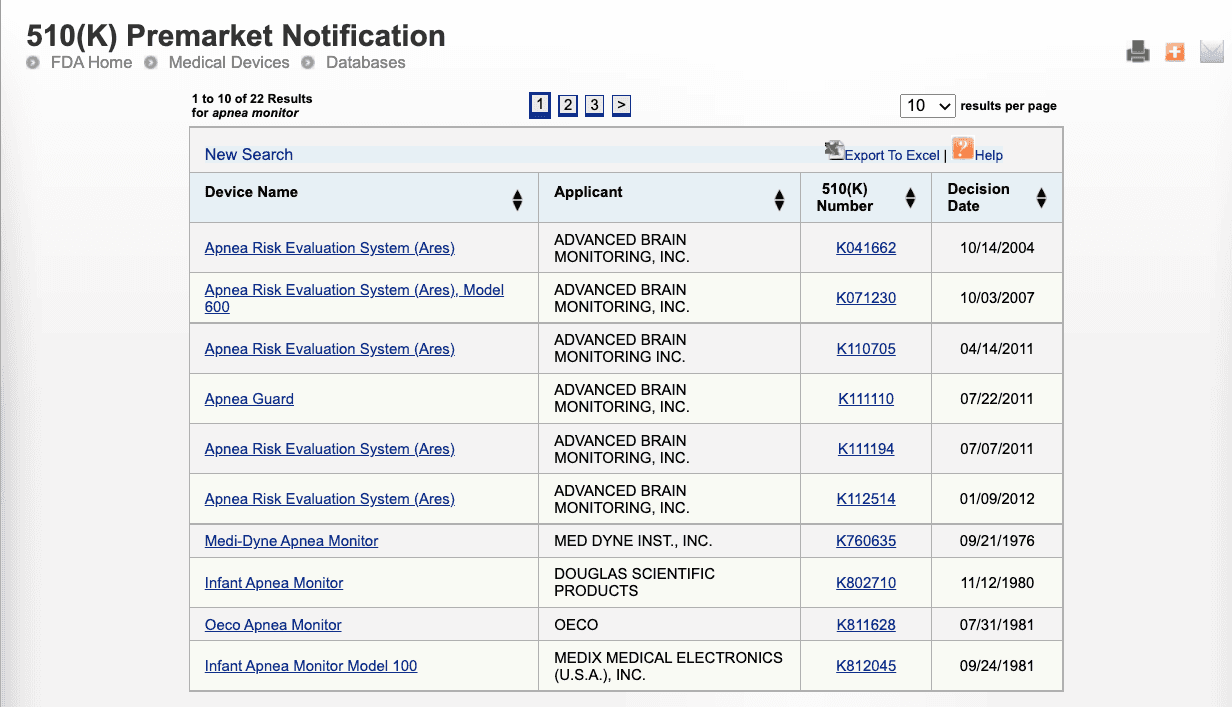
Click search to view the list of devices that have a name apnea on them. This lists the devices with the keyword Apnea in them with decision dates and applicant information, all of which is valuable information that you can use for your own application.
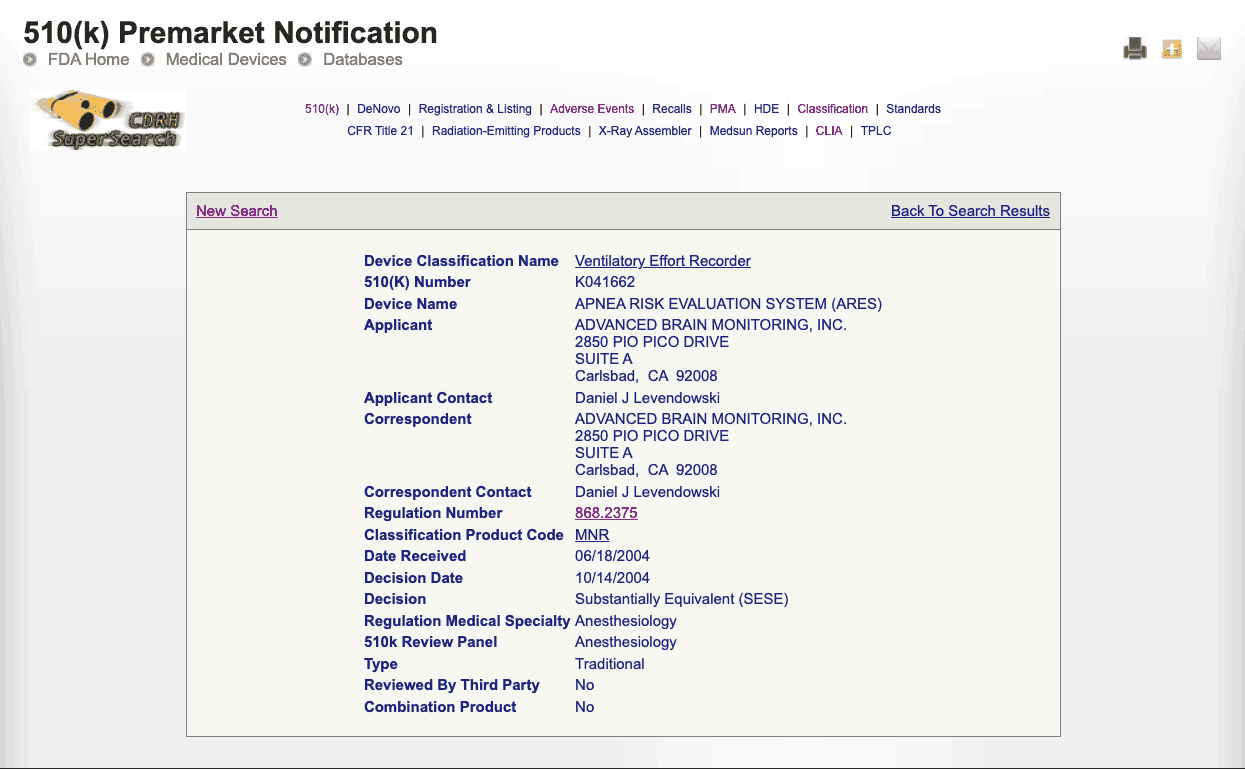
Click on the device name to see the regulatory attributes such as medical speciality, common name, and regulation number, and text. Scan the list and note down the products and regulatory attributes that look relevant. Also, click on the 510(k) Number and note down additional details for further use in the advanced search.
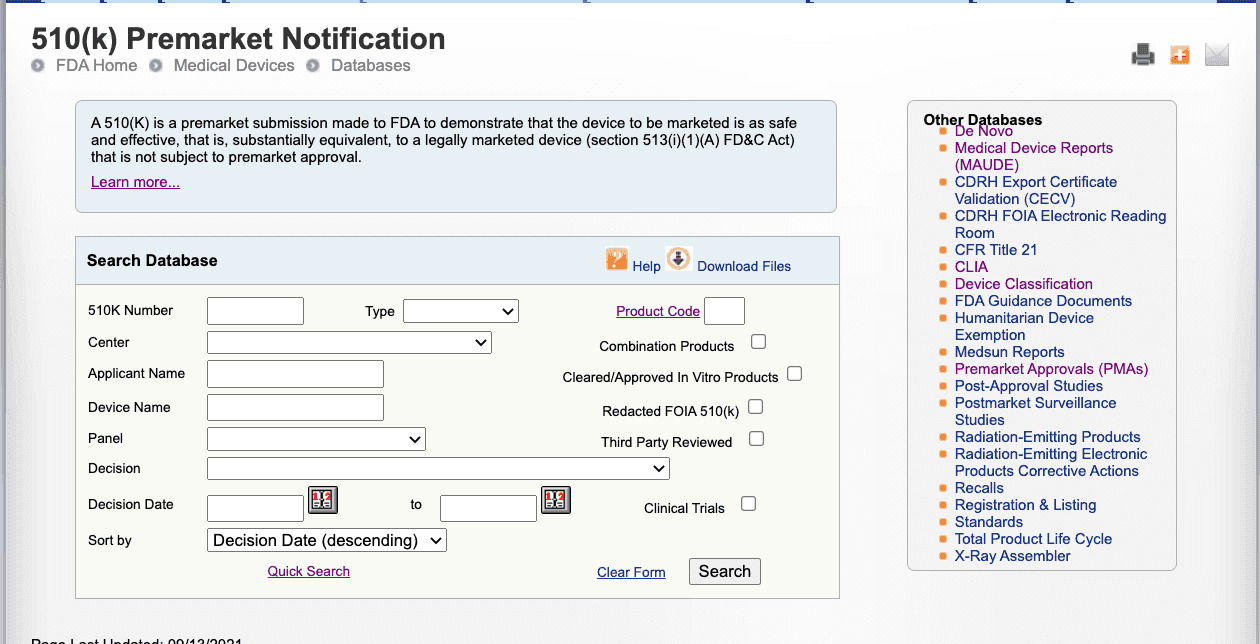
The advanced search screen (shown below) is helpful when you already have key data on your competitors such as their 510(k) number or three letter product code. It is also helpful if are already confident the device in questions is similar to yours.
Assimilate results, compare relevant attributes, and rank order search results
Now that you have collected product code, regulations, and summary documents of possible similar devices, it is time to compare them with your technology, indications, mode of action and components. As well as, identifying the one that is closest to your device.This will then help you identify the applicable tests and guidance documents relevant to your device. This will inform your broader regulatory strategy and therefore your budget and timelines to go to market.
Managing Regulatory strategy and ongoing regulatory Intelligence
Regulatory assessment is a task that medical device companies must complete regularly in order to stay current with the latest regulatory changes. It is paramount that regulatory teams maintain a proactive approach to understand and assess the impact of changes to regulations, standards, and new guidance documents published by FDA, as well as device recalls of similar devices or changes in the product category.
Methods used today include manually searching in guidance documents, standards databases, recalls and MAUDE reports databases that FDA has made available for public search. A slightly more sophisticated approach is subscribing to a plethora of industry journals, attending conferences, subscribing to FDA newsletters and notifications to stay up-to-date.
The problem with this approach is regulatory affairs specialists and leaders end up consuming a lot of data from disparate data sources and struggle to make sense out of it all. This requires an audit of these sources to narrow down the information relevant to your device. Then, you need to assess what you just learned and recommend a course of action. With the number of rapid changes occurring every day, these approaches quickly become unmanageable.
Limitations of the current process to search in FDA 510(k) Database:
Whether you are using keywords-based search or competitor data for regulatory strategy, these searches can take hours. You also risk the possibility of omission due to the time the number of records that need to be reviewed.
Evaluating competitor technology could be helpful. However, elements such as technological characteristics, materials, and mode of action do not always equal a device that you can use to show substantial equivalence.
Therefore, finding the right substantially equivalent device seldom involves a straightforward strategy. Instead, it is the result of hours of careful preparation of a combination of keywords. This list that can identify a list of devices already in the market and then doing a couple of passes to compile a list of devices and technology.
The siloed process of passively managing regulatory attributes, strategy, and search criteria while spending hours scouring the internet is time-consuming and prone to error. Reviewing the regulatory attributes alongside the technology profile will help you identify the ideal devices to use to establish substantial equivalence.
A better way to manage regulatory search and intelligence
What if there is a way to combine all the steps mentioned above and streamline the entire process? A tool that can allow you to preview and analyze critical regulatory attributes for specific devices that are ranked by relevance based on your list of keywords.
What if you were are able to pull the regulatory updates from health authorities related to you products? What if your team could leverage a regulatory intelligence system that understands important attributes and can also notify you when changes occur.
As busy regulatory specialists, leads, and executives in medical device companies, your strength lies in your ability to make great decisions substantiated by data points. Therefore access to relevant regulatory information curated without noise can save hours and sometimes days and it can be a long-term competitive driver of medical device innovation.