

Here is a list of questions we should understand:
What is a predicate device and how to claim substantial equivalence to a proposed device?
Types of devices needing risk management for 510(k) submissions?
Risk management requirements for a Traditional 510(k)?
Where can one find 510(k) risk management requirements?
How can manufacturers demonstrate device safety?
Key factors for documenting risks before submitting 510(k)?
Contents of software related documentation?
What are the contents of risk related documentation?
Using Substantial equivalence to demonstrate risk management in the 510(k) submission.
A predicate device is a legally marketed device subjected to premarket approval. We can demonstrate Substantial Equivalence by comparing the applicant’s device to a cleared predicate device, if:
it was cleared through the 510(k) process
it was legally marketed prior to May 28, 1976 (pre-amendments device)
it was originally on the U.S. market as a Class III device (Premarket approval) and later down-classified to Class II or I
it is a 510(k)-exempt device
We can demonstrate substantial equivalence if predicate device has:
The same intended use as the predicate; and
the same technological characteristics as the predicate
The same intended use as the predicate; and
different technological characteristics and the information submitted to FDA and
does not raise new questions of safety and effectiveness; and
demonstrates that the device is at least as safe and effective as the legally marketed device.
Same technical characteristics can be:
The design
Energy used or delivered
Materials of construction
Performance
Safety
Effectiveness
Labeling
Biocompatibility
Environmental conditions
Storage/ transport
operating
If the new device has some different technological characteristics the differences must not raise questions of safety or effectiveness when you’re testing the device as safe and effective as the predicate device. However, if testing is done in accordance with FDA recognized standard then certificate of compliance to that standard is sufficient for the submission, and global test information or test data is not required.
A simple way to show substantial equivalence is to create a comparison table including:
Technological specifications mentioned below:
Intended use, Indications for use
Target population, Anatomical sites, where used (hospital, home, etc.)
Energy used and/or delivered
Human factors, Design, Performance, Standards met, Materials, Biocompatibility, Compatibility with environment and other devices
Sterility, Electrical safety, Mechanical safety, Chemical safety, Thermal safety, Radiation safety
A Narrative discussing similarities and differences between new and predicate device
Devices requiring risk management for 510(k) submission.
Risk management requirements are only for devices that contain software.
Abbreviated 510(k)s generally require declarations of conformity and risk management documents
Risk management requirements for a Traditional 510(k).
Only embedded software, driven by or standalone software and devices with software component must include Hazard Identification and Risk Assessment in 510(k)s.
When there is insufficient information to create general controls, special controls provide reasonable assurance of safety and effectiveness. It assures FDA that hazards related to device development process were subject to controls and mitigations.
You should include Risk Control Verification and Validation requirements under
Packaging Validation
• Sterilization Validation
• Biocompatibility
• Software Verification Validation
• Electrical Safety and EMC
• Bench/Pre-Clinical Performance Testing
• Animal Performance Testing
• Clinical Performance Testing
Risks to Users and Patients are addressed in the Instructions for Use (IFU) as warnings, contraindications, and precautions (typically because of usability studies and hazard analysis).
Risk-Benefit Analysis is required in Special 510(k)s, De Novo applications, Humanitarian Device Exemptions, and PMAs. It is not required for traditional 510(k).
Where can one find 510(k) risk management requirements?
Design validation-software validation and risk analysis (21 CFR 820.30) FDA and EU CE Marking compliance- recognize ISO 14971 standard:
FDA recognizes ISO 14971:2019
EU recognizes EN ISO 14971:2019 (differences include omission of Annexes, decoupling of standards to MDR regs)
Best practices to address risks in 510(k) submissions
Look for appropriate Special Controls Guidance Docs in the FDA Guidance Document
Use Guidance Documents for Controls and Risk Management Requirements
Examine the guidance and determine which standards, testing, and hazard/risk analyses are appropriate.
Ensure extensive testing
How to demonstrate device safety?
The GHTF/SG1-N11:2008 in 10.0 Risk Analysis and Control Summary states that “The STED [Summary Technical Documentation] should contain a summary of the risks identified during the risk analysis process and how these risks have been controlled to an acceptable level. Preferably, this risk analysis should be based on recognized standards and be part of the manufacturer’s risk management plan.”
Factors to consider regarding risks before submitting 510(k)?
Human factors:
“Medical device manufacturers are required to follow FDA’s Human Factors guidance and regulations to help ensure safe use of these devices.” – CDRH website
Recognized standards include ISO 14971 risk management standard, IEC 62366 (joint ISO/IEC standard applying to all devices), IEC 60601-1-6 and AAMI HE75. To comply with standards,
Manufacturers will have to develop usability specifications for devices
Manufacturers should explore usability issues as part of the risk management process in compliance with standards
Include Usability evaluations as part of design validations
The FDA requires a summary of these actions as part of a human factors dossier while submitting pre-market information, which is explained in detail below:
Description of device user interface
Description of user interaction with UI (use scenarios)
Device, Training, Labeling, IFU
intended users, uses, use environments and training
Summary of known use problems from:
Predecessor devices
Similar devices
Analysis of hazards and risks associated with the use of the device
Summary of preliminary analyses and evaluations
Description and categorization of critical tasks
Details of human factors validation testing
Description of test participants (minimum of 15 in each user population)
Description of test scenarios
Moderator guideline
Training offered
Test results
Test results summary
Conclusion
Content of Software-related risk documentation.
In the 510(k) submission, medical device manufacturers must:
show they identified hazards appropriately and managed risks effectively and
provide traceability to link design, implementation, testing, and risk management.
The following information is picked from the Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices, May 11, 2005
Device Hazard Analysis: Tabular description of identified hardware and software hazards, including severity assessment and mitigations.
Traceability Analysis: Traceability among requirements, specifications, identified hazards and mitigations, and Verification and Validation testing.
When performing a hazard analysis, it is recommended that you address all foreseeable hazards, including those resulting from intentional or inadvertent misuse of the device.
The risk documentation for software can be in the form of an extract of the software-related items from a comprehensive risk management document, such as the Risk Management Summary described in ISO 14971. In this format, each line item should include:
identification of the hazardous event
the severity of the hazard i.e., level of concern
cause(s) of the hazard i.e., device Hazard analysis
method of control (e.g., alarm, hardware design)
corrective measures taken, including an explanation of the aspects of the device design/requirements, that eliminate, reduce, or warn of a hazardous event; and
verification that the method of control was implemented correctly.
Software Description
Software Requirements Specification (SRS)
Architecture Design Chart
Software Design Specification (SDS)
Traceability Analysis
Software Development Environment Description
Configuration management & maintenance plan
Verification/ validation Documentation
Revision Level History
Unresolved anomalies
Impact on safety or effectiveness discussion
Rationale for accepting
It is recommended to base your estimation of risk for your Software Device on the severity of the hazard resulting from failure, assuming that the failure will occur. You need to use risk identification and control techniques described in consensus standards such as ISO 14971.
Documenting risk in 510(k)
After analyzing the previous material, it is evident that ISO 14971 has identified a method for providing the information necessary by the FDA, namely the traceability summary required in Clause 3.5 of the risk management file.
The information can be provided in a document that is displayed in the GHTF guidance, GHTF/SG3/N15R8 Implementation of risk management principles and activities within a Quality Management System Annex C.
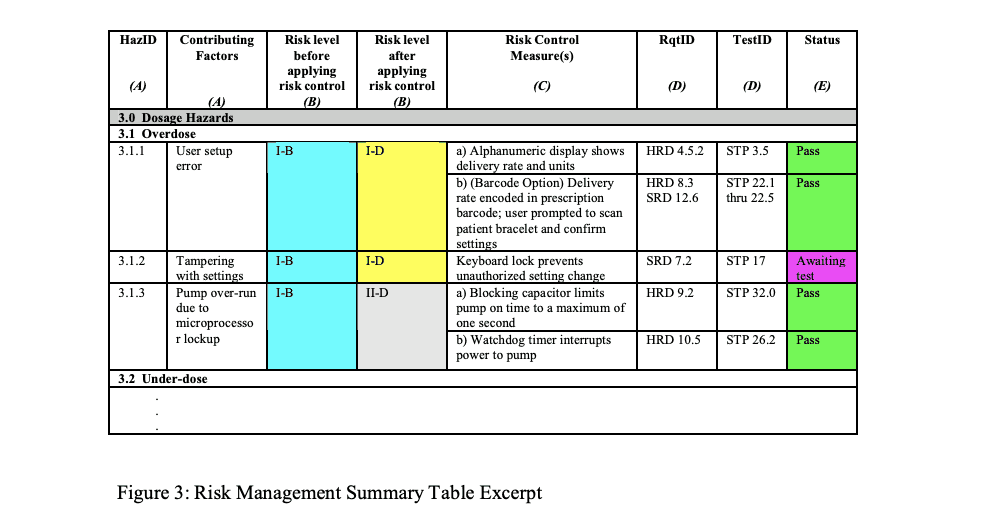
Example of Risk Management Summary Table
Risk Traceability Summary
You can add a column with reference to the source document listing each hazard and additional information to improve the use of Risk Traceability Summary, for example:
A Human Factors or Usability study that identifies hazards
A standard that identifies a hazard and possibly a risk control method
MAUDE database identified hazard from predicate device
Complaint file from previous similar product
Risk Information in 510(k)
Provide a copy of the Risk Chart, which specifies the Risk Acceptability levels for the product in the submission, and include copies of the definitions of Severity Levels and the Probability of Occurrence Levels.
Take care of probability levels to ensure there is evidence to support the quantified levels and the ranges do not overlap.
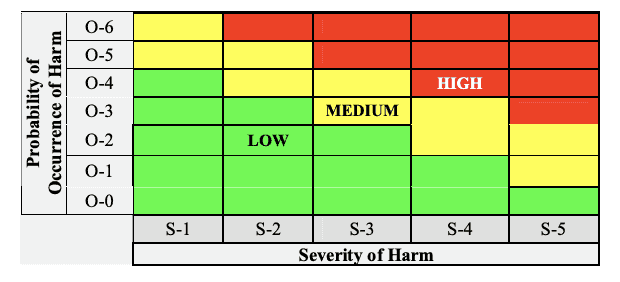
Risk chart communicating risk management activities
Conclusion
Managing 510(k) is a tedious process, requiring data from various teams and several elements of test strategy. However, you can still navigate the process of organizing all the data in a simple yet comprehensive manner. It is important to remember that FDA reviewers can only review so much detail, and therefore randomly put together irrelevant data can cause delays in receiving approval. Therefore too much information can negatively impact 510(k) review timelines. It is a manufacturer’s responsibility to submit data accurately and present it in an easily comprehensible format. FDA can have many questions after the application is submitted, and they need to be responded promptly by going back to the source of data and responding to FDA to compress your approval cycle.
Leveraging Regulatory Management Technology (RIM) to build your medical device technical files and 510(k) submissions will help you avoid inconsistency, maintain clarity for managing submission requirements helping you streamline only those processes and parts of of section of your document that need additional detail.
About Essenvia
Essenvia is a regulatory lifecycle management solution to help you optimize the entire regulatory operations process. Our Regulatory Management Software solutions include an innovative approach to building your pre-market submissions like 510(k) and MDR/ IVDR as well as post-market submissions like Regulatory Change Assessment and Post Market Surveillance. Our solution reduces errors, streamlines regulatory process, and guarantees faster regulatory clearance for your medical device.
Schedule a demo now to find out how Essenvia can help you with your regulatory submissions.